- 移动端


广州卿泽生物科技有限公司
6 年
手机商铺
- NaN
- 0.8999999999999999
- 1.9
- 0.8999999999999999
- 3.9
推荐产品



公司新闻/正文
【线粒体翻译】 |eIF5A“超修饰”解密:p53如何通过线粒体翻译调控衰老细胞免疫监视
363 人阅读发布时间:2025-09-25 11:47
导读:
~~衰老细胞的“翻译加速器”之谜~~
细胞衰老伴随增殖停滞,其分泌的衰老相关分泌表型(SASP)因子既可清除癌前细胞,也可能促进慢性炎症。

本研究首次发现:衰老细胞蛋白合成速率显著高于增殖细胞,且不依赖于致癌基因激活。通过CRISPR筛选,锁定真核翻译起始因子eIF5A为关键调控者——其特有的羟腐胺赖氨酸修饰(hypusination)依赖多胺代谢产物亚精胺,是维持衰老细胞高翻译率的核心开关。这一发现串联起p53、线粒体翻译与免疫监视的三重调控网络,为抗衰老及癌症干预提供新视角。
文章索引:
标题:P53-dependent hypusination of eIF5A affects mitochondrial translation and senescence immune surveillance.
发表期刊:Nature Communication.
发表时间:2024.08
作者团队:德国癌症研究中心 Fabricio Loayza-Puch团队
IF:15.7
DOI:10.1038/s41467-024-51901-w.
核心故事线
衰老细胞需要超高翻译速率→eIF5A是维持该状态的关键→其功能依赖hypusination修饰→该修饰由p53-SMOX-亚精胺轴调控→最终影响线粒体翻译和免疫清除
研究结果
(1)衰老细胞中的蛋白质合成增加
首先,作者通过OP-Puro标记技术(图1a),系统量化了衰老细胞的蛋白合成速率。在癌基因诱导衰老模型中,BJ-Ras-ER、IMR-90-Ras-ER和TIG3-BRAF-ER细胞的OP-Puro掺入率均显著高于增殖对照组(图1b-d:),证实衰老细胞蛋白合成速率平均提升2-3倍。这一现象不仅限于癌基因激活:治疗诱导衰老(TIS)模型中,经多柔比星(DOXO)或喜树碱(CPT)处理的A549、HCT116、HeLa和MCF7细胞同样显示合成速率升高(图1e)。复制性衰老模型(IMR-90细胞,群体倍增PD=49)进一步验证了全局趋势(图1f),表明高翻译速率是衰老细胞的普遍特征。
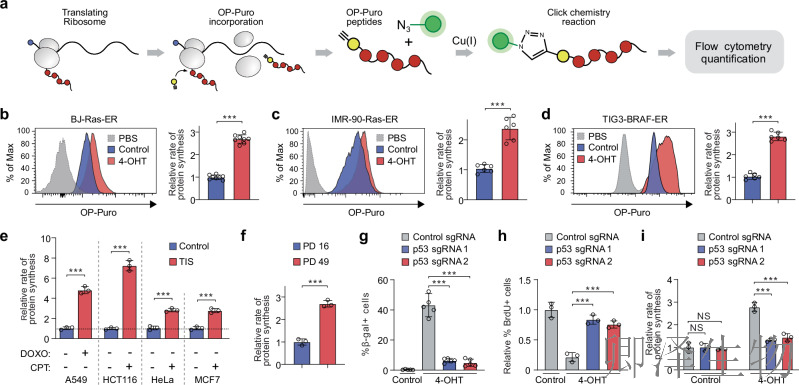
值得注意的是,p53敲除实验揭示该效应不依赖致癌基因:sgRNA靶向p53后,衰老标志SA-β-gal表达显著降低(图1g),细胞突破增殖停滞(BrdU掺入率升高,图1h),但OP-Puro掺入率在衰老细胞中降至增殖水平(图1i),证明衰老相关高翻译需p53参与而非致癌信号驱动。
综上,上述结果确立衰老细胞的核心代谢重塑——蛋白合成速率全局性升高,为后续机制解析奠定基础。
(2)在细胞衰老过程中,需要eIF5A维持较高的蛋白质合成速率
通过CRISPR筛选(图2a)锁定eIF5A为衰老细胞高翻译速率的特异性依赖因子(图2b);Western blot验证(图2c)证实eIF5A高效敲除;功能上,eIF5A缺失使衰老细胞OP-Puro掺入率降低50%(图2d),但不影响细胞周期停滞(BrdU掺入率不变,图2e)、SA-β-gal阳性率(图2f)或p16/p21表达(图2g);时序干预模型(图2h-i)进一步揭示:在已建立的衰老细胞中敲低eIF5A,仍可显著抑制翻译(图2j)且不逆转生长停滞(图2k)。
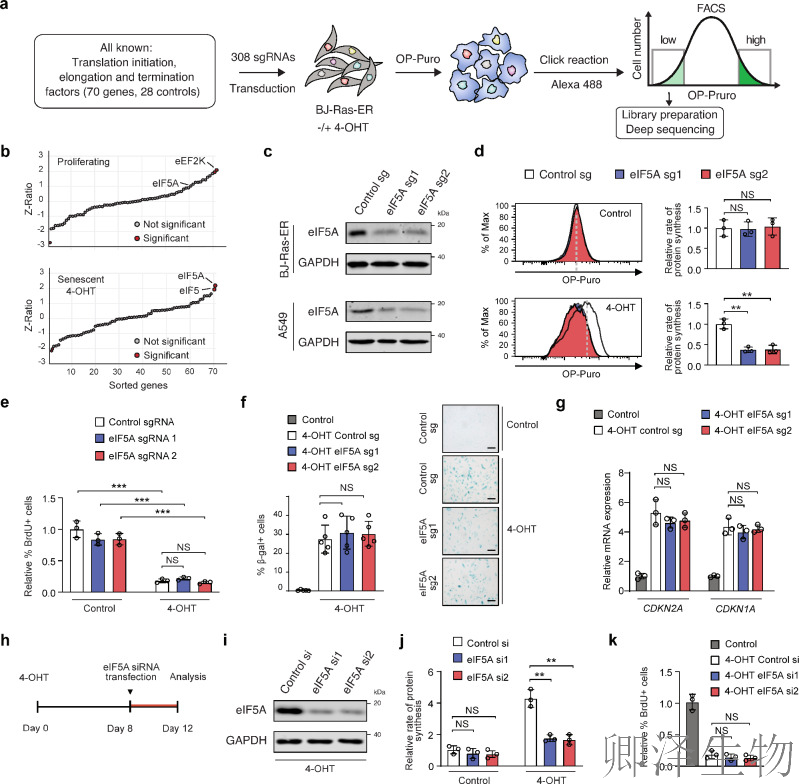
综上,eIF5A是维持衰老细胞高翻译状态的特异性分子开关,其功能独立于经典衰老信号通路。
(3)p53在细胞衰老过程中调节多胺合成途径组分的表达
eIF5A 是一已知携带翻译后修饰——羟腐胺化的蛋白质。在人类细胞中,多胺亚精胺是eIF5A中一个保守赖氨酸残基发生羟腐胺修饰的底物,这一过程包括两个连续步骤,分别由脱氧羟腐胺合成酶(DHPS)和脱氧羟腐胺羟化酶(DOHH)催化(图3a)。用GC7(一种DHPS抑制剂)处理的OIS BJ-Ras-ER细胞在维持羟腐胺修饰水平方面比增殖细胞更敏感(图3b-c)。与这些观察结果一致,GC7处理或DHPS/DOHH敲除仅在衰老细胞中降低了蛋白质合成速率(图3d),表明多胺代谢在调控衰老细胞升高的蛋白质合成速率方面发挥了作用。
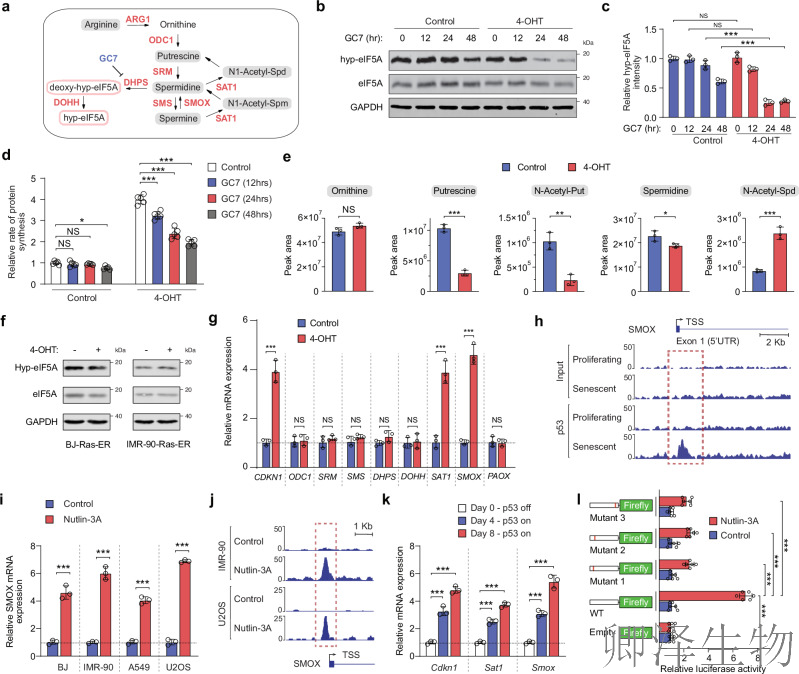
为了研究多胺在OIS过程中对eIF5A的羟腐胺修饰所起的作用,作者使用液相色谱/质谱(LC/MS)进行了代谢组学分析,以测定增殖性和衰老的BJ-Ras-ER细胞中多胺代谢物的水平。衰老细胞中腐胺和N-乙酰腐胺显著降低,但亚精胺维持稳态,保障eIF5A修饰底物(图3e)。与亚精胺的可比水平一致,OIS或TIS中eIF5A的羟腐胺修饰未受到影响(图3f)。这些发现表明,尽管腐胺水平较低,衰老细胞中可能存在一种补偿机制以维持亚精胺和eIF5A的羟腐胺修饰水平。
为了探讨这种可能性,作者评估了参与多胺生物合成或分解的关键酶的表达。qRT-PCR分析显示,衰老细胞中亚精胺/精胺N1-乙酰转移酶1(SAT1)和精胺氧化酶(SMOX)mRNA的表达增加(图3g),而多胺代谢通路中其他酶的表达则未发生变化。
SAT1已被证明是p53的直接靶点,而SMOX的转录调控仅在缺氧条件下被表征。在衰老细胞中,SAT1和SMOX可能受到p53的直接调控,使精胺再循环回亚精胺,维持eIF5A的hypusination水平。ChIP-seq数据发现p53直接结合SMOX启动子(图3h)。
此外,作者发现p53激活剂Nutlin3A使SMOX表达升高(图3i-j), IMR-90和U2OS细胞中p53与SMOX启动子的结合增加(图3j)。此外,p53修复诱导表达致癌Ras(H-RasG12D)的小鼠肝祖细胞中Smox和Sat1 mRNA的表达(图3k)。将SMOX启动子克隆到一个荧光素酶报告基因中,,并观察到p53稳定后的强劲激活(图31)。
综上所述,研究结果表明,SAT1和SMOX是p53的直接靶点,并表明这两种酶在细胞衰老过程中维持亚精胺和eIF5A活性水平的作用。
(4)SMOX维持eIF5A的精胺酸化修饰并促进OIS期间的蛋白质合成
精氨酸是多胺前体鸟氨酸合成的主要底物(图4a)。作者推测p53的靶点SAT1和SMOX可能通过重塑多胺代谢通路,在衰老细胞中维持亚精胺水平并促进蛋白合成。为验证此假设,在敲低SAT1或SMOX的衰老BJ-Ras-ER细胞中进行¹³C精氨酸同位素示踪。结果显示:SAT1敲低不影响¹³C标记的亚精胺水平,但SMOX敲低显著降低亚精胺水平(图4b),表明SMOX对维持衰老细胞亚精胺池至关重要。同时,N-乙酰亚精胺和总精胺水平同步下降(图4b)。
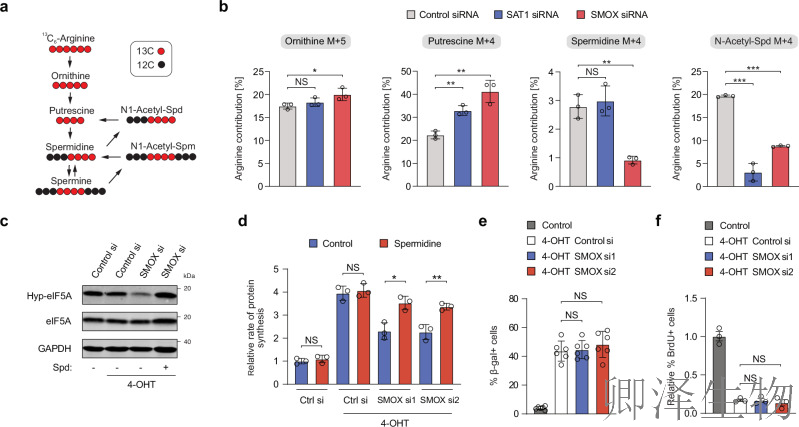
为评估SMOX对eIF5A羟腐胺化修饰的影响,作者采用siRNA敲低或MDL 72527来抑制SMOX。发现SMOX敲低降低eIF5A修饰水平,但外源亚精胺可挽救此效应(图4c)。OP-Puro检测显示蛋白合成速率下降(图4d),但细胞生长停滞不受影响(图4e,f)。
综上,p53靶向的SMOX是维持eIF5A修饰及衰老细胞高翻译速率的核心因子。
(5)eIF5A调节衰老细胞中线粒体核糖体蛋白的翻译
为了评估eIF5A活性如何调控衰老细胞中的蛋白质合成,作者对增殖状态的BJ-Ras-ER细胞和OIS BJ-Ras-ER细胞进行了整体蛋白质组分析,并对这些细胞进行了GC7处理。
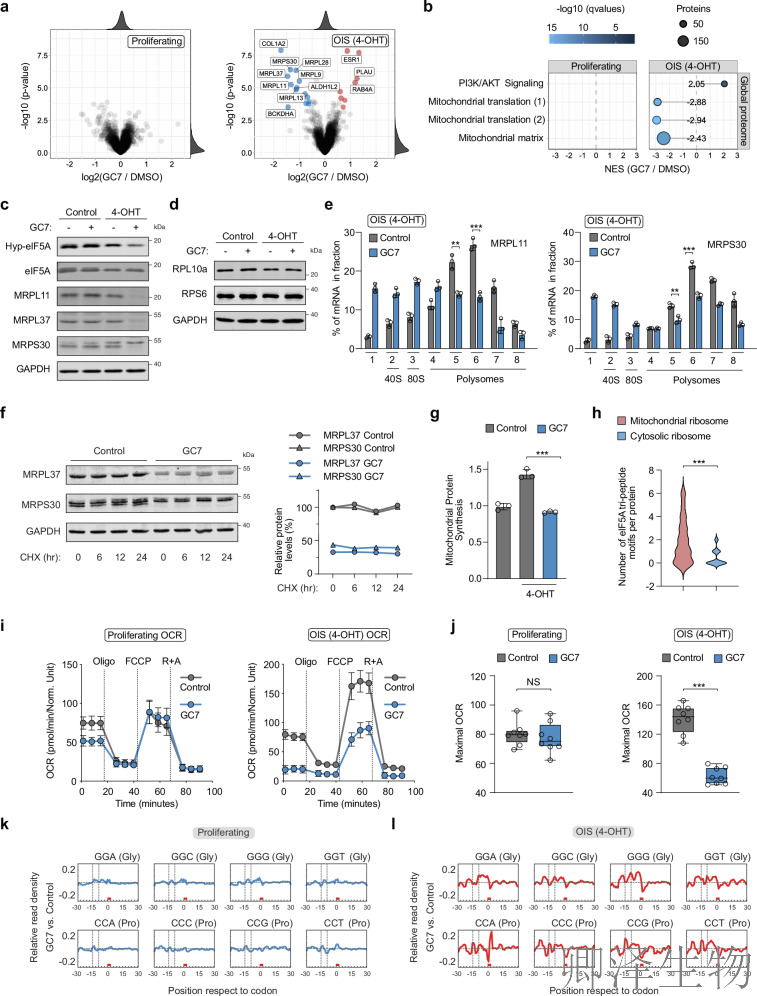
通过全局蛋白组学分析(图5a),GC7抑制eIF5A修饰后,衰老细胞中线粒体核糖体蛋白(如MRPL37/MRPS30)显著下调,而胞质核糖体蛋白无变化,GSEA证实"线粒体翻译"通路特异性受抑(图5b);Western blot验证线粒体核糖体蛋白表达降低(图5c-d),多聚核糖体分析显示其mRNA翻译效率下降50%(图5e),CHX追踪排除降解影响(图5f);线粒体特异性OP-Puro掺入率降低(图5g),伴随呼吸功能崩溃(基础OCR↓, 最大呼吸↓, 图5i-j);机制上,线粒体核糖体蛋白mRNA富含eIF5A依赖的脯氨酸/甘氨酸三联体(图5h),核糖体谱证实GC7处理后在脯氨酸(A位)和甘氨酸(E位)密码子处显著停滞(图5k-l)。该数据阐明eIF5A通过缓解特定位点核糖体卡顿维持线粒体翻译稳态。
(6)抑制eIF5A hypusination影响SASP的表达,损害体内致癌衰老细胞的免疫监视
既往研究表明SASP(衰老相关分泌表型)依赖线粒体功能。线粒体翻译缺陷会增加活性氧(ROS)产生,进而抑制SASP表达。
通过抗体芯片分析发现,衰老细胞条件培养基中CXCL1、IL-8等炎性因子显著增加(图6a),而GC7(eIF5A修饰抑制剂)处理显著降低CXCL1、IL-8、PTX3和THBS1水平(图6a,b)。qRT-PCR证实eIF5A抑制会降低促炎因子mRNA转录(图6c)。证明eIF5A通过调控线粒体翻译促进促炎SASP表达。
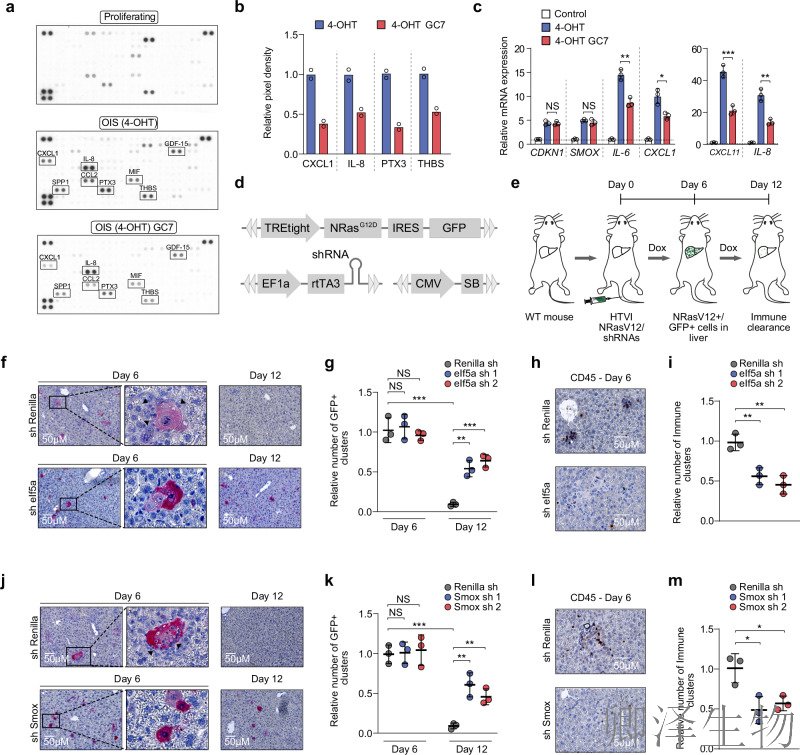
在体内模型中,通过水动力尾静脉注射(HTVI)将携带致癌基因NrasG12D和靶向eIf5a/Smox的shRNA载体导入小鼠肝脏(图6d,e)。注射6天后,各组均出现SA-β-gal阳性衰老肝细胞(图6f,g,j,k);注射12天后,对照shRenilla组衰老细胞被清除,而sh-eIf5a或sh-Smox组衰老细胞滞留(图6f,g,j,k),且CD45+免疫细胞浸润减少(图6h,i,l,m)。表明eIF5A和多胺回收对SASP介导的体内免疫清除至关重要。
研究结论
这项研究结果为eIF5A在调节衰老细胞线粒体核糖体蛋白翻译中的关键作用提供了证据。这种调节对线粒体功能和促炎SASP的表达有显著影响。本研究结果的意义延伸到更广泛的衰老过程背景下,强调了eIF5A及其与多胺相互作用的重要性。这些关于eIF5A参与线粒体核糖体蛋白翻译的分子机制的见解,为肿瘤发生过程中调节年龄相关疾病和增强免疫监测的潜在治疗靶点提供了线索。
欢迎致电了解详情或咨询!
广州卿泽生物科技有限公司
地址:广州市黄埔区伴河路96号一栋三层321房
电话:18925086102(微信同号)