- 移动端


广州卿泽生物科技有限公司
6 年
手机商铺
- NaN
- 0.8999999999999999
- 1.9
- 0.8999999999999999
- 3.9
推荐产品



公司新闻/正文
【核糖体碰撞-经典研究】 UV致死元凶非DNA损伤?核糖体毒性应激终解密!
197 人阅读发布时间:2025-09-23 15:39
导读:
虽然紫外线(UV)辐射会损伤DNA,引发DNA损伤反应(DDR),但它也会损伤RNA,导致整个转录组范围内的核糖体碰撞,并引发核糖体毒性应激反应(RSR)。然而,这些通路在决定细胞命运中的相对作用、发生时序及调控机制仍不清楚。

本研究通过对紫外线照射后分子和细胞变化的综合分析表明,核糖体毒性应激反应而非DNA损伤应答介导了紫外线依赖性的程序性细胞死亡。
文章索引:
标题:The ribotoxic stress response drives UV-mediated cell death.
发表期刊:Cell.
发表时间:2024.07
作者团队:约翰霍普金斯大学医学院 Rachel Green 团队
IF:42.5
DOI:10.1016/j.cell.2024.05.018.
研究结果
(1)对紫外线辐射的即时早期反应主要是由核糖体介导的信号传导
首先,作者通过多组学技术揭示了UV辐射后核糖体介导的信号在早期响应中的主导地位。
示意图(图A)展示了核糖体碰撞激活ZAK和GCN2的分子机制;随后,免疫印迹实验(图B)证实ZAK抑制剂可特异性阻断UV诱导的p38/JNK磷酸化,而GCN2抑制剂则阻断eIF2α磷酸化;活细胞成像(图C)进一步显示UV剂量依赖的细胞命运分化——低剂量诱导G2期阻滞,高剂量触发凋亡;蔗糖梯度离心(图D)直接捕捉到UV照射1分钟内核糖体碰撞体(二聚体/三聚体)的爆发式形成;
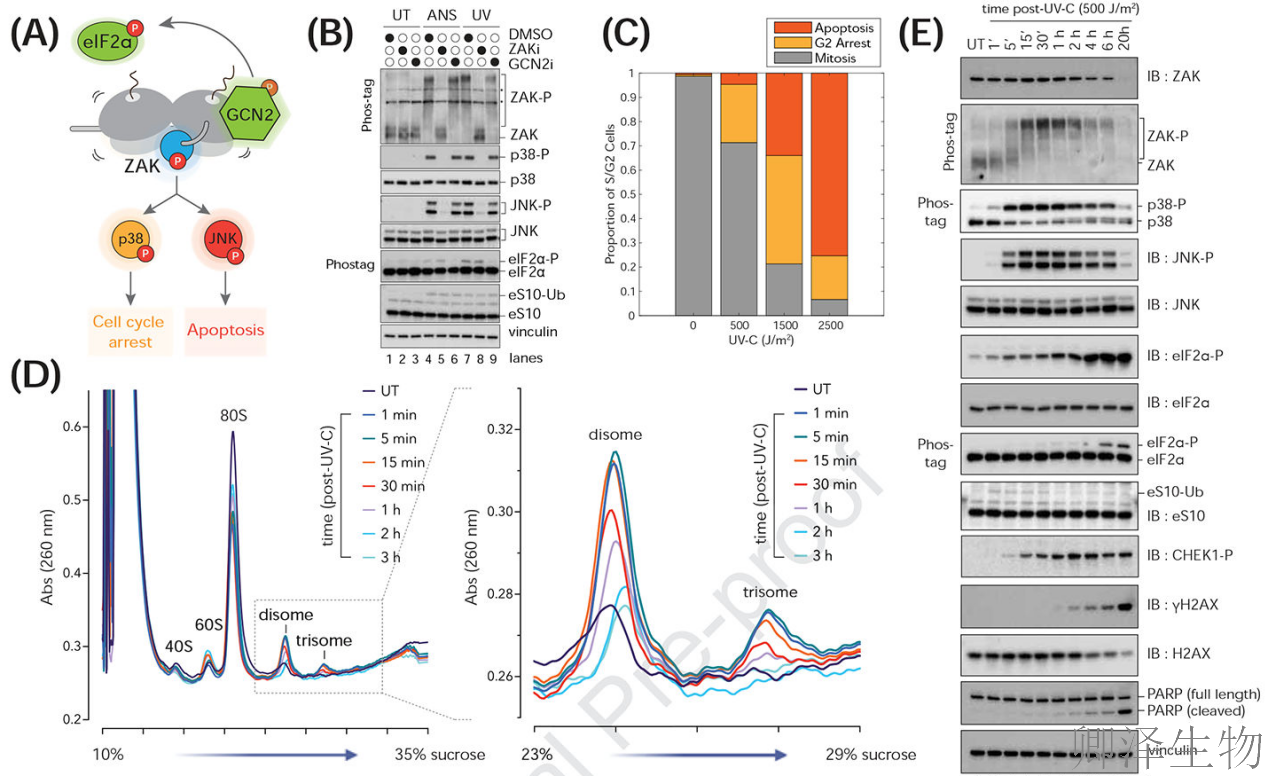
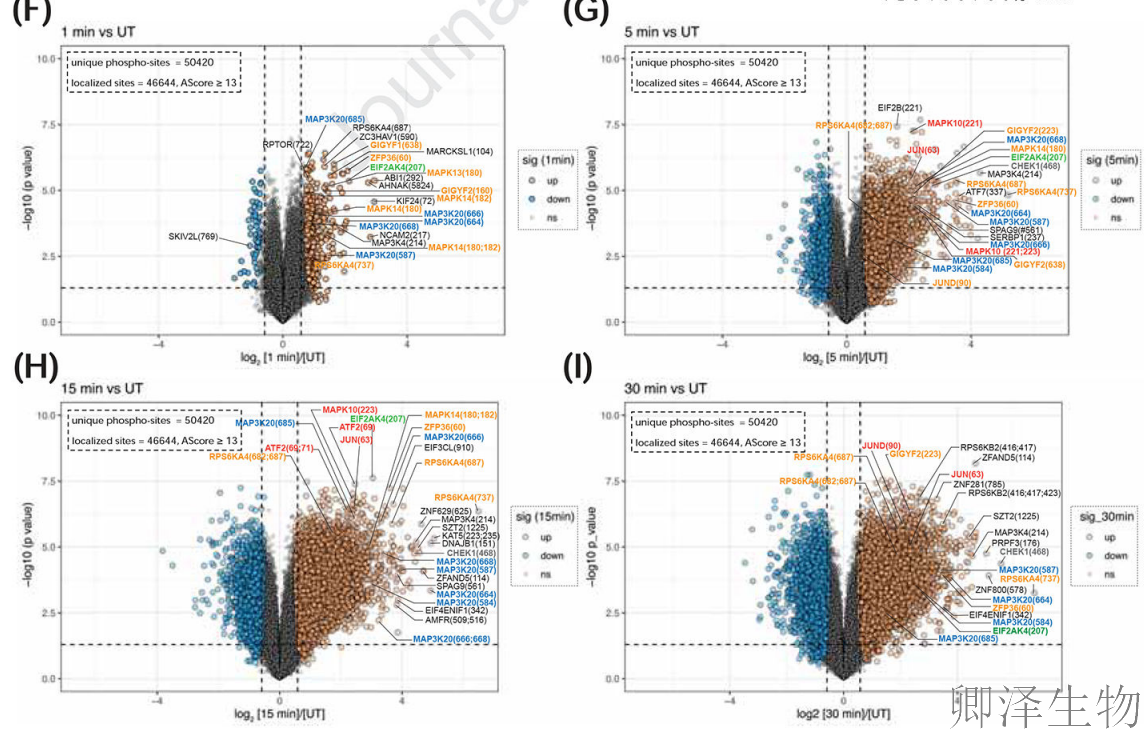
时间分辨免疫印迹(图E)揭示ZAK-p38-JNK信号级联在5分钟内迅速激活,远早于DDR通路;最后,磷酸化蛋白质组的火山图分析(图F-I)系统描绘了RSR通路激酶的早期主导性激活,其中1分钟即有399个磷酸位点显著变化,且ZAK底物MAP2K4等早于DDR效应分子。
(2)ZAK和GCN2定义了细胞响应紫外线应激时的早期磷酸化蛋白质组
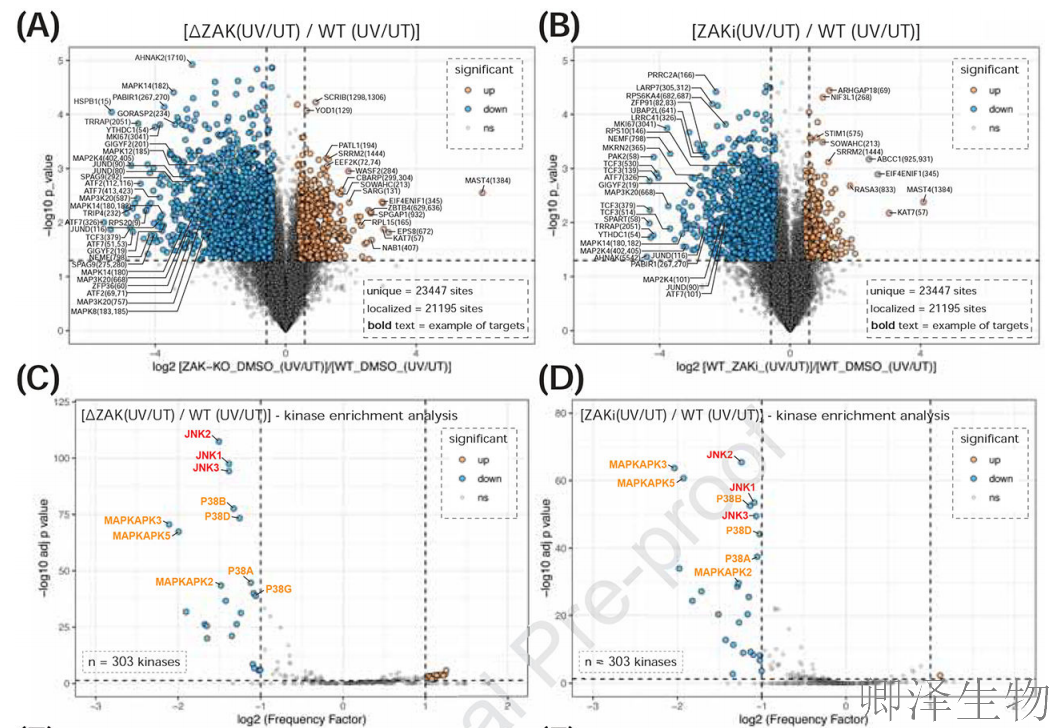
接着,作者研究了ZAK和GCN2在紫外线处理后对磷酸化蛋白质组重塑的贡献。紫外线(UV)引发的核糖体毒性应激中,ZAK和GCN2如同细胞信号乐团的指挥。
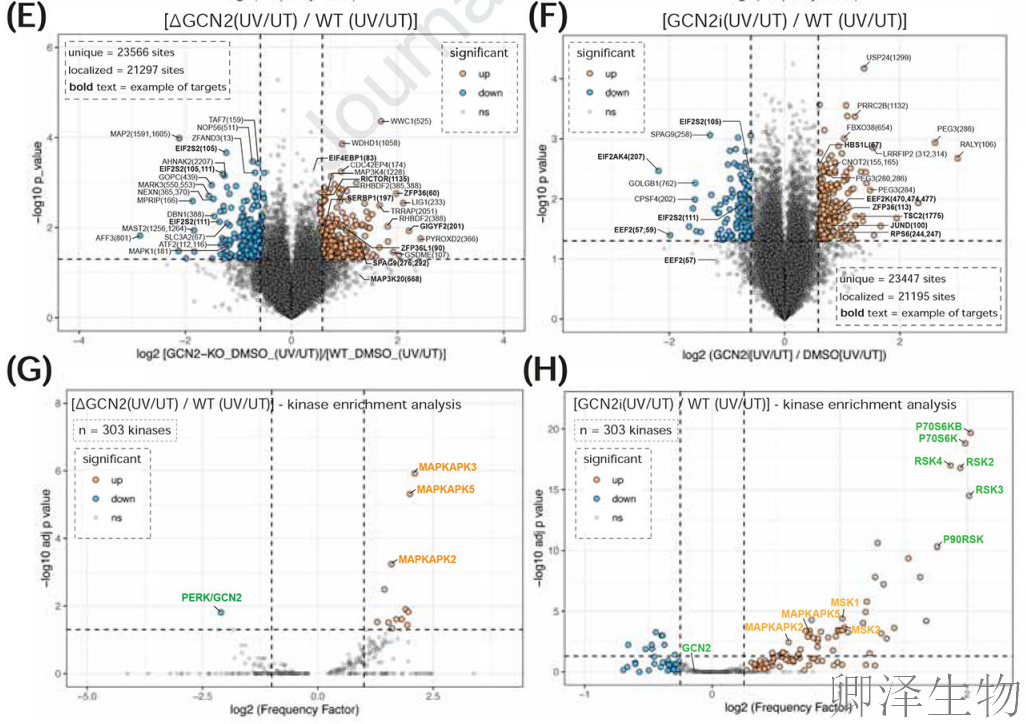
磷酸化组全景分析(图A-B)发现:ZAK缺失使2487个磷酸位点沉默(占全组10.5%),其中p38/JNK激活环磷酸化完全消失(如MAP2K4-S257/T261),而激酶网络分析(图C-D)进一步证实p38效应器MAPKAPK2/3/5活性暴跌;令人意外的是,GCN2缺失(图E-F)虽使221个磷酸位点下调(如eIF2α-S51),却触发mTOR信号失控——核糖体蛋白RPS6磷酸化异常升高,这种双向失衡(图G-H)最终解释了一个致命循环:当GCN2失效时,翻译过载加剧核糖体碰撞,反而通过激活ZAK-JNK通路将细胞推向凋亡深渊。
(3)细胞对紫外线的反应中,细胞死亡是由ZAK介导的,而不是通过DDR通路
随后,作者发现当紫外线(UV)照射细胞时,ZAK介导的核糖体应激响应(RSR)而非DNA损伤响应(DDR)才是凋亡的真正推手。
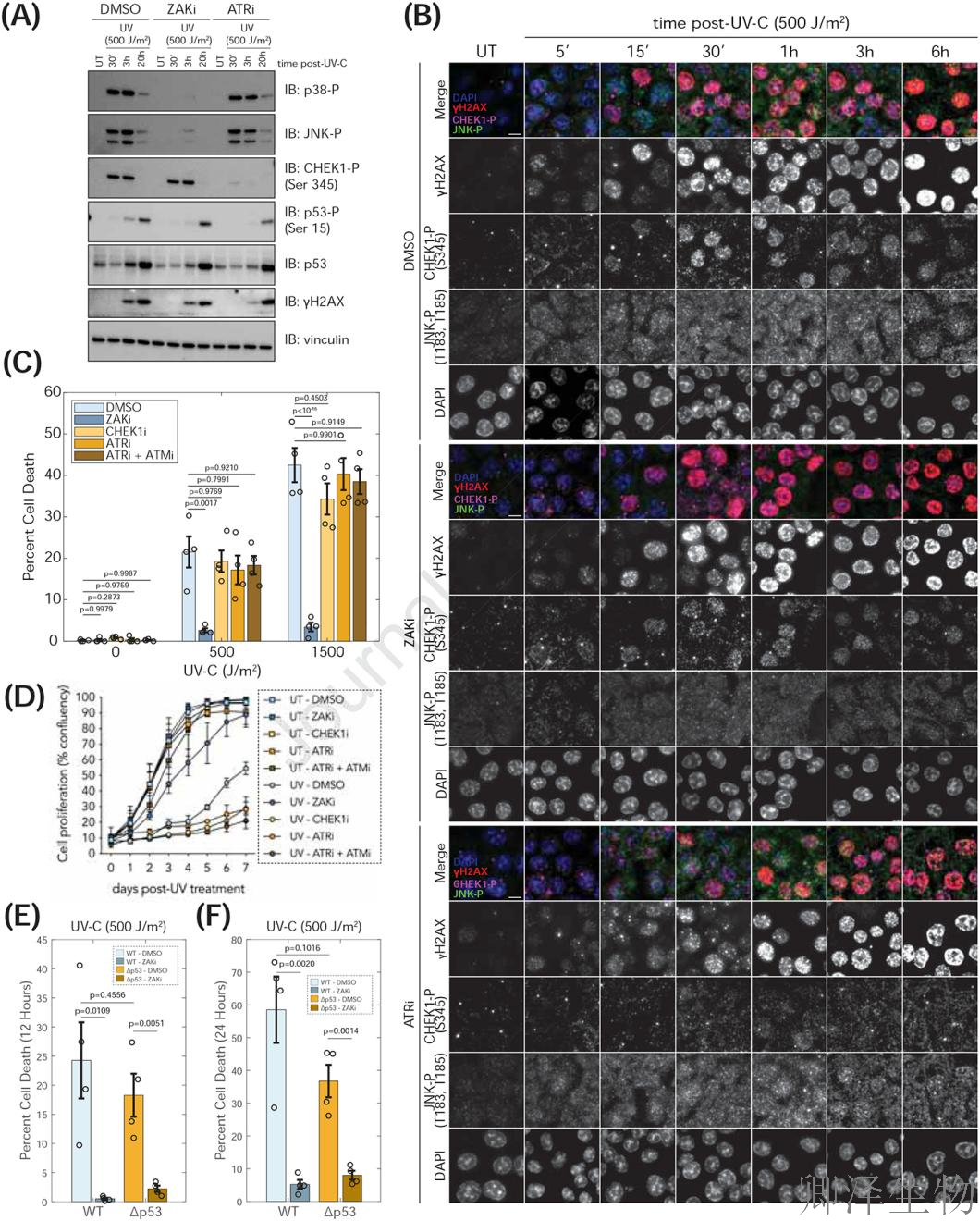
免疫印迹(图3A)显示ZAK抑制剂选择性阻断p38/JNK磷酸化却不影响CHEK1激活,而ATR抑制剂的作用恰恰相反;这种通路独立性在细胞水平得到印证(图3B):所有细胞均出现JNK磷酸化(绿色),但仅S期细胞激活CHEK1(红色)。更关键的是,凋亡检测(图3C)证明只有ZAKi能完全挽救UV诱导的死亡,即使联合阻断ATR/ATM也无效;长期培养(图3D)进一步揭示ZAKi赋予细胞显著的增殖优势,而DDR抑制剂反而加剧损伤。最震撼的证据来自p53缺失细胞(图3E-F):尽管经典凋亡通路被切断,ZAKi仍能完全阻断死亡,证明p53非依赖性凋亡完全由ZAK主导。
(4)GCN2通过限制核糖体负荷来防止ZAK介导的细胞死亡
磷酸化蛋白质组数据对GCN2在核糖体毒性应激中的作用提出了两个有力的预测:(1)GCN2通过磷酸化eIF2α并抑制mTOR活性来阻断翻译起始;(2)在缺乏GCN2的情况下,受损mRNA上翻译起始增加,导致碰撞核糖体的积累并过度激活RSR。
预测GCN2的缺失会增加翻译起始,从而导致更多的碰撞。事实上,在UV-C处理后,ΔGCN2细胞中二聚体、三聚体和四聚体的比例增加,同时单体的比例下降(图4A);那在没有GCN2的情况下,碰撞核糖体的积累是否会通过增加ZAK活性而使RSR过度激活?
用UV-C处理MCF10a WT和ΔGCN2细胞,发现在ΔGCN2细胞中,eS10泛素化始终保持升高,表明碰撞核糖体持续积聚(图4B)。p38的激活表现出较温和的增加(图4B)。
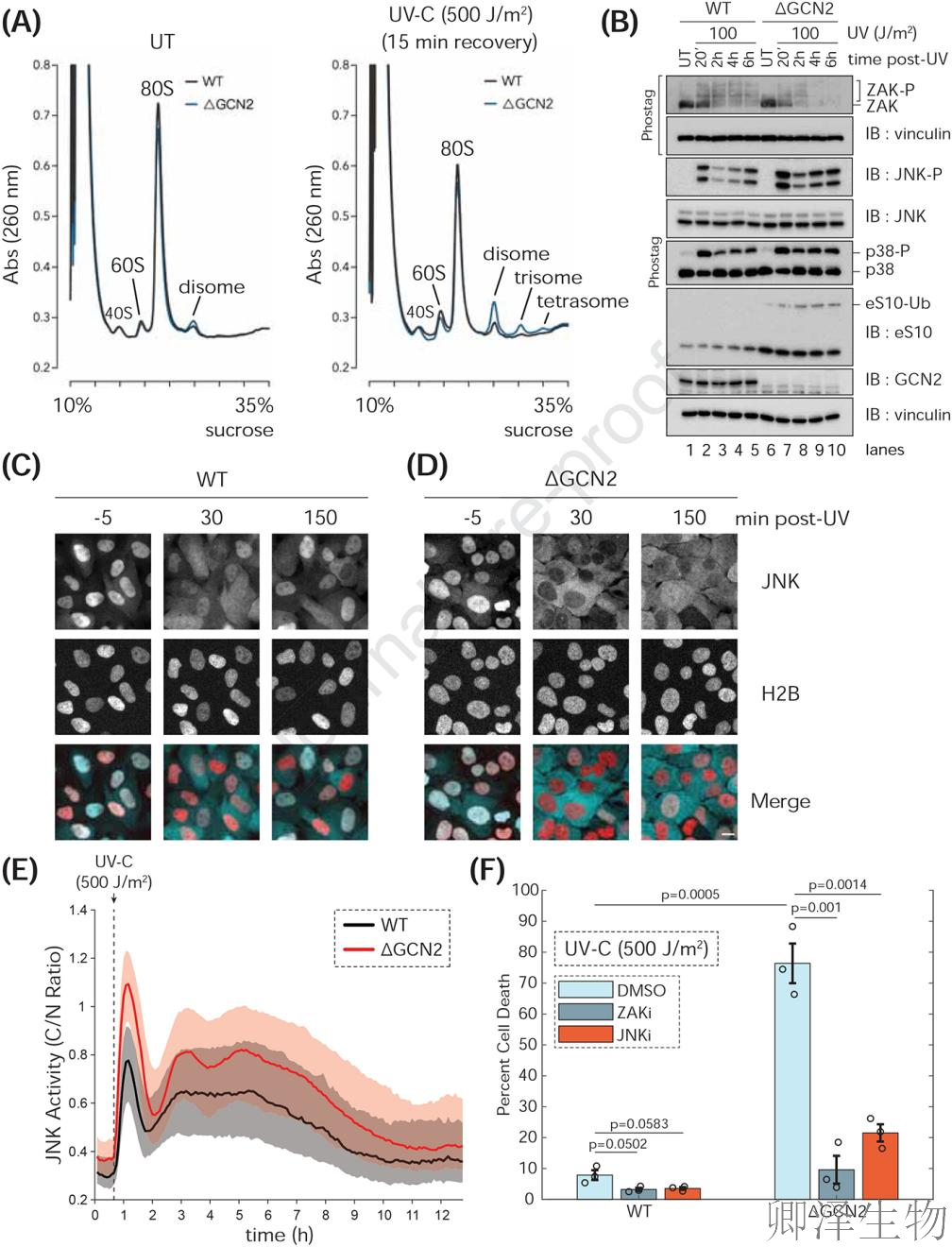
为了更好地跟踪JNK动态,作者在WT和ΔGCN2细胞中对核毒性应激反应的JNK KTR进行了活单细胞成像。在未经处理的细胞中,JNK KTR主要位于核内,表明基础活性较低。UV-C处理后,JNK-KTR转移到细胞质中,表明活性增加(图4C-D)。JNK活性在20分钟达到峰值,在1小时时下降,然后在几个小时内保持中间脉动状态(图4E)。虽然两种细胞类型表现出相似的JNK动态,但ΔGCN2细胞显示出更高的JNK总体活性。
假设ΔGCN2细胞中JNK活性的增加会导致细胞凋亡的增加。作者对WT和ΔGCN2细胞进行了活细胞成像,并测量了紫外线处理后12小时的细胞死亡情况。看到未经处理的细胞几乎没有细胞死亡,紫外处理后WT细胞凋亡百分比略有增加,用ZAKi或JNKi预处理可以逆转这种情况(图4F)。相反,ΔGCN2细胞在UV处理后表现出明显更高的凋亡水平,也被ZAKi或JNKi逆转(图4F)。这些结果支持一个模型,其中GCN2激活限制碰撞核糖体在受损mRNA上的积累,并通过减弱ZAK介导的JNK信号传导来限制细胞凋亡。
(5)ZAK自磷酸化调节核糖体解离及其后续降解
当核糖体碰撞激活ZAK时,其C端核糖体结合域(RBR)的⁶⁵⁶DSGFSS⁶⁶¹降解基序成为决定命运的开关——磷酸化组分析(图B-C)发现34个磷酸位点形成三簇动态响应,其中集群2(S587/S660/S668)在UV刺激15分钟达峰并密集包围降解基序(图D箭头);这触发了分子级联:免疫沉淀质谱(图E)显示碰撞诱导β-TrCP结合暴增,而蔗糖梯度实验(图F)证实集群2磷酸化突变体(S-A)顽固结合核糖体,野生型则快速解离;最终降解路径锁定(图G-K):CRL1抑制剂MLN4924和蛋白酶体抑制剂硼替佐米完全阻断ZAK降解,但集群2突变体对此无响应,揭示自磷酸化-β-TrCP募集-泛素化降解是ZAK的制动程序。
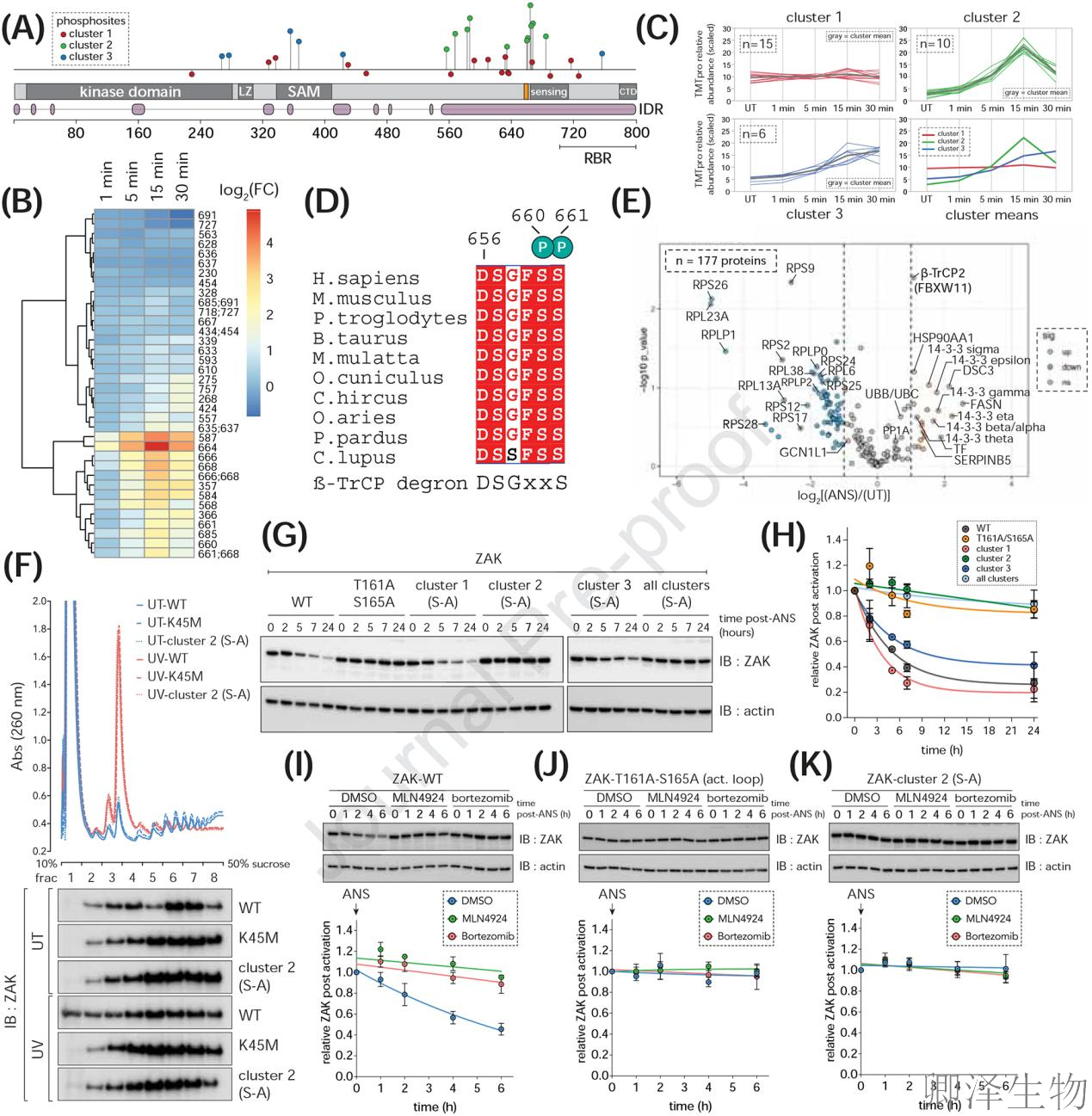
大致形成了如下的机制闭环:

(6)ZAK降解在持续性核糖体毒性应激条件下限制细胞凋亡并诱导耐受性
作者推测程序化的ZAK降解调控细胞命运,为此系统比较了ZAK磷酸化突变体(Cluster 1/2/3及全突变体)与ΔZAK、野生型(WT)及激酶死亡突变体(K45M, T161A-S165A)的活性(图6A)。关键发现显示:Cluster 2磷酸化位点突变体(S-A)在基础状态下即异常活跃,Phos-tag免疫印迹(图6A,第7/9 vs 第3泳道)证实其迁移率改变,伴随JNK磷酸化显著升高,揭示Cluster 2位点组成性抑制基础核糖体碰撞引发的JNK信号泄露;而UV刺激下,所有突变体的ZAK/p38/JNK磷酸化水平与野生型无差异(图6A),证明激酶结构域(而非磷酸化集群)主导核心应激响应。
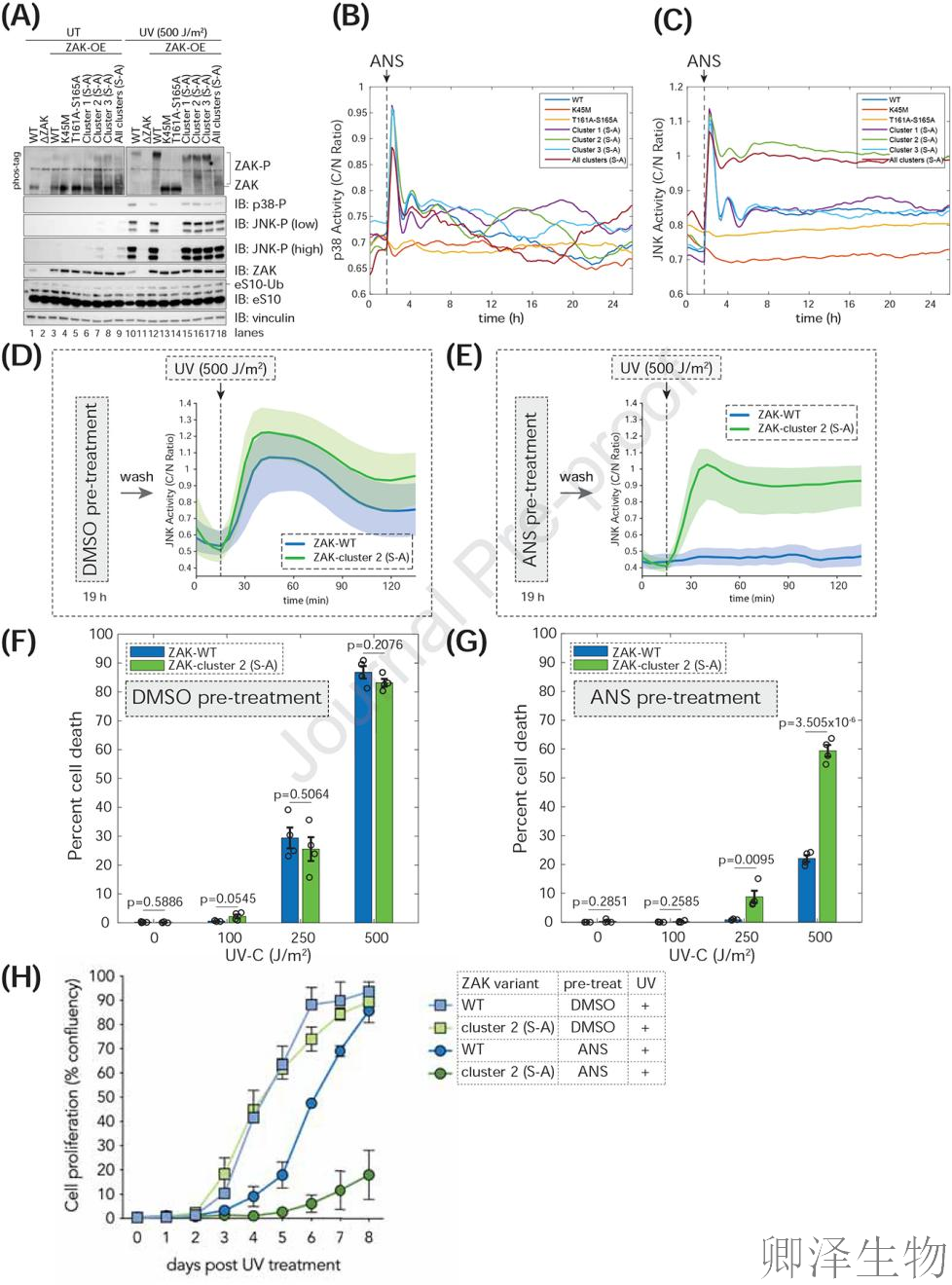
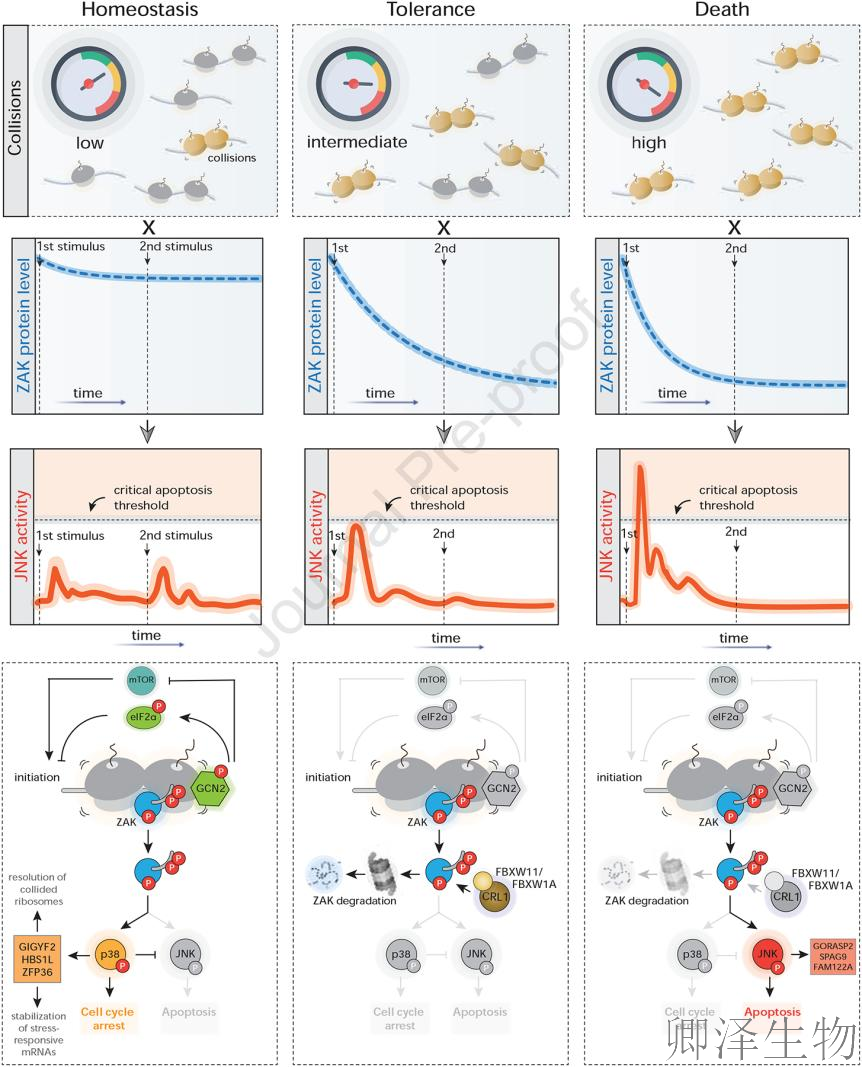
通过p38/JNK激酶转位报告系统(KTRs)动态监测发现:Cluster 2磷酸化位点突变体(S-A)在核糖体应激下呈现JNK信号持续滞留(野生型呈现脉冲式激活后衰减,而突变体亢进24小时)(图6C);当细胞经历低剂量ANS预处理时,野生型ZAK通过降解建立耐受态(后续UV刺激无JNK响应),而突变体因降解失灵导致JNK剧烈激活(图6E);耐受态下的野生型抵抗中剂量UV杀伤(凋亡率<20%),而突变体因"分子刹车失灵"凋亡率飙升至60%(图6G),长期增殖能力同步崩溃(图6H)。这些证据链共同确证:ZAK降解是细胞应对持续性应激的"分子缓冲器":其功能完整时,细胞获得修复时间窗;功能缺陷时,则坠入凋亡深渊。
研究结论
这项研究揭示出:核糖体碰撞的强度和持续时间充当细胞命运决策的分子调谐器:低水平碰撞(如短暂UV暴露)激活p38通路诱导G2期阻滞,维持稳态;中等水平碰撞触发ZAK自磷酸化降解,建立耐受态以缓冲持续应激;高水平碰撞(如高强度UV)导致ZAK水平激增和JNK持续活化,突破凋亡阈值。这一机制确立ZAK为细胞应激响应的核心剂量传感器,直接将核糖体损伤的动力学转化为生存、耐受或死亡的生物学结局。
欢迎致电了解详情或咨询!
广州卿泽生物科技有限公司
地址:广州市黄埔区伴河路96号一栋三层321房
电话:18925086102(微信同号)